29 Regulatory Submission
The culmination of years of research, billions of dollars in investment (see Chapter 6), and thousands of patients’ participation comes down to a single question: will regulators approve the drug? The regulatory submission is where sponsors present their case: the accumulated evidence from preclinical studies and clinical trials that, they hope, demonstrates the product is safe and effective.
Different types of applications serve different products and circumstances:
- New Drug Application (NDA): the primary vehicle for small-molecule drugs in the United States, containing complete data on chemistry, manufacturing, preclinical studies, and clinical trials
- Biologics License Application (BLA): for biological products such as proteins, vaccines, and cellular therapies, following a similar structure but with additional manufacturing details specific to biologics
- Marketing Authorization Application (MAA): the equivalent submission in the European Union, submitted centrally or through national procedures
- Abbreviated New Drug Application (ANDA): facilitates approval of generic drugs by relying on reference products
- Biosimilar applications: demonstrate similarity to an approved reference biologic
- Supplements and variations: allow sponsors to modify existing approvals for new indications or manufacturing changes
International harmonization has established a standardized format for regulatory submissions known as the Common Technical Document (CTD). The CTD organizes information into five distinct modules, as summarized in Table 29.1.
| Module | Name | Content Description |
|---|---|---|
| 1 | Regional Administrative | Forms, labeling, and jurisdiction-specific materials |
| 2 | Summaries & Overviews | Integrated quality, nonclinical, and clinical summaries |
| 3 | Quality (CMC) | Chemistry, Manufacturing, and Controls documentation |
| 4 | Nonclinical | Reports from toxicology and pharmacology studies |
| 5 | Clinical | Clinical study reports and related data materials |
This standardized modularity allows sponsors to prepare a single “core dossier” (comprising Modules 2 through 5) for submission to multiple global regulatory agencies simultaneously. Only Module 1 requires region-specific adaptation, significantly streamlining the process of bringing a new therapy to multiple international markets.
29.1 Building the Submission
Assembling a major regulatory application requires close coordination across multiple specialized functions. Regulatory affairs teams lead the effort, managing the submission strategy and maintaining direct communication with agencies to ensure every legal and technical requirement is met. The clinical development group manages the compilation and interpretation of all clinical data, while CMC specialists document the manufacturing and quality controls that guarantee product stability. Simultaneously, nonclinical researchers provide foundational toxicology data, and medical writers synthesize these disparate streams into clear summaries that help agency reviewers navigate thousands of pages of technical information. The final submission is packaged as an electronic CTD (eCTD) for secure transmission to regulatory authorities worldwide.
29.2 Pre-Submission Interactions
Smart sponsors do not simply submit applications and hope for the best. They engage with regulatory agencies throughout development and especially before major submissions.
Pre-IND meetings occur before first-in-human studies to clarify the data needed to support clinical development. End-of-Phase 2 meetings are a key checkpoint for discussing Phase III plans, where sponsors present their design and evidentiary standards for agency feedback. Finally, Pre-NDA/BLA meetings (or Pre-MAA in Europe) are conducted before the major submission to confirm that the dossier is complete and free of critical unresolved issues.
These meetings, typically conducted as formal written exchanges or in-person discussions, help align sponsor and agency expectations. Different meeting types have different timelines and objectives, as summarized in Table 29.3. Discovering a major issue before submission is far better than discovering it during review.
29.3 Expedited Regulatory Pathways
In response to the urgent need for treatments for serious conditions, regulatory agencies have established specific pathways designed to accelerate the development and review of promising therapies. These pathways do not lower the evidentiary standard for safety or efficacy; rather, they intensify the interaction between the sponsor and the regulator, and in some cases allow for the use of surrogate endpoints reasonably likely to predict clinical benefit.
Fast Track designation is intended for drugs that treat serious conditions and fill an unmet medical need. This status allows for more frequent meetings with the FDA and the potential for rolling review, where the sponsor submits completed sections of the application before the entire dossier is finalized. Breakthrough Therapy is a more intensive designation for drugs where preliminary clinical evidence indicates the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint. Accelerated Approval allows approval based on a surrogate endpoint or an intermediate clinical endpoint that is reasonably likely to predict clinical benefit. Finally, Priority Review represents a commitment by the FDA to complete review within six months for drugs that would provide significant improvements in safety or effectiveness.
Table 29.2 contrasts these distinct mechanisms. Note that a single drug program may utilize multiple pathways simultaneously (e.g., a Breakthrough Therapy that receives Accelerated Approval and Priority Review).
| Designation | Criteria | Key Benefit |
|---|---|---|
| Fast Track | Serious condition + unmet need | Rolling review, frequent communication |
| Breakthrough | Serious condition + substantial improvement | Intensive guidance, organizational commitment |
| Accelerated Approval | Serious condition + surrogate endpoint | Approval based on surrogate (requires confirmation) |
| Priority Review | Significant improvement | 6-month review clock (vs 10 months) |
| Meeting Type | Purpose | Examples | FDA Response Goal |
|---|---|---|---|
| Type A | Critical path issues; Stalled development | Dispute resolution; Clinical hold discussion | 30 days |
| Type B | Key development milestones | Pre-IND, End-of-Phase 2, Pre-NDA/BLA | 60 days |
| Type C | General guidance | Protocol design review; New indication discussion | 75 days |
29.4 The Review Process
Once submitted, applications enter formal review shown in Figure 29.1.
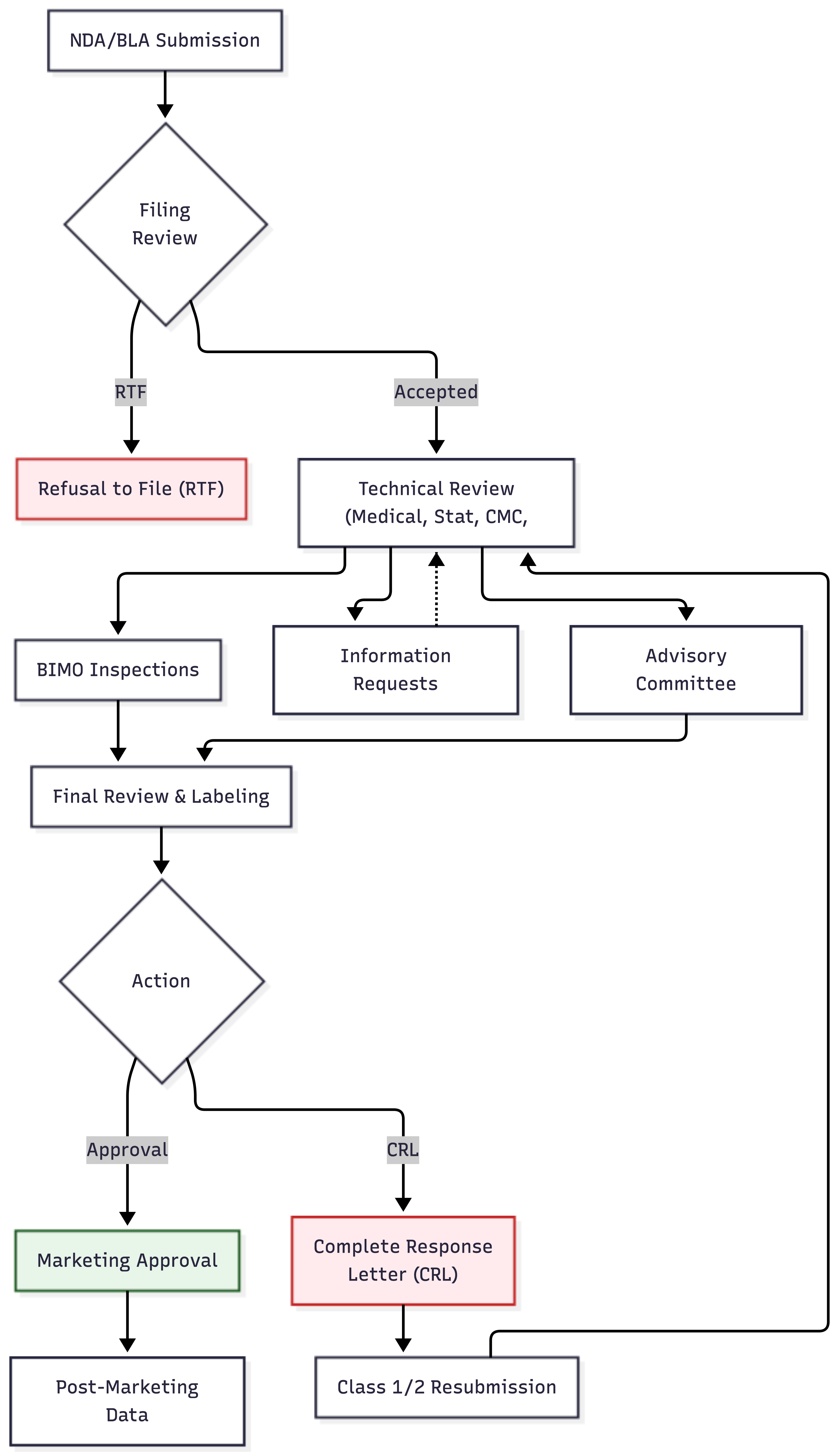
In the United States, FDA first determines whether the submission is complete (that is, whether it contains enough information to permit substantive review). Applications deemed incomplete may be refused to file and returned without review.
If accepted, the application enters a review queue. Review timelines are governed by user fee goals, performance targets established under the Prescription Drug User Fee Act (PDUFA). Standard review typically takes 10-12 months from acceptance; priority review may be completed in 6 months (U.S. Food and Drug Administration 2014).
Review is multidisciplinary: medical officers evaluate clinical efficacy and safety; pharmacologists assess pharmacokinetics; chemists review manufacturing; statisticians verify the analyses; toxicologists examine nonclinical safety.
29.5 Bioresearch Monitoring (BIMO) Inspections
Parallel to the scientific review of the data, the FDA conducts on-site inspections through its Bioresearch Monitoring (BIMO) program to verify the integrity of the data and compliance with regulations. Positive data are worthless if the agency cannot trust that they were collected accurately and ethically.
BIMO inspections typically target clinical sites, the sponsor, and the involved Institutional Review Boards. At clinical sites, inspectors compare source documents against the data submitted in the case report forms, looking for discrepancies or evidence of fraud. Sponsor inspections evaluate the oversight of the trial, checking monitoring reports, data management systems, and safety reporting processes. IRB inspections ensure that ethical oversight was maintained appropriately throughout the study.
Significant findings during a BIMO inspection can result in a Form 483 (listing observations) or a Warning Letter (U.S. Food and Drug Administration 2024a). In severe cases, the FDA may invoke the “Application Integrity Policy,” pausing the review until the data’s reliability can be re-established. Consequently, “inspection readiness” is a continuous activity for clinical operations teams, not just a pre-submission drill.
Reviewers may issue information requests, which require the sponsor to clarify or supplement the submission. Sponsors must respond promptly to avoid delays.
For novel or complex applications, FDA may convene an Advisory Committee, a panel of outside experts who discuss the application in a public meeting and vote on approval recommendations. While advisory committee recommendations are not binding, FDA usually follows them.
29.6 The Decision
At the conclusion of review, FDA issues an action letter.
An approval letter authorizes marketing. The product may now be sold, but usually with specific labeling, potentially with post-marketing requirements or commitments, and subject to ongoing safety monitoring.
A complete response letter (CRL) indicates that the application cannot be approved in its current form. The letter specifies deficiencies that must be addressed. Sponsors may then submit a response, addressing each deficiency. Multiple review cycles may be needed before approval is achieved, or the sponsor may ultimately abandon the product.
Similar decision frameworks exist in other jurisdictions: Europe uses marketing authorization and requests for supplementary information; Japan and other markets have analogous processes.
29.7 Single-Trial Substantial Evidence
The traditional interpretation of “substantial evidence” (introduced at Chapter 9) requires replication across at least two adequate and well-controlled trials. But the statistical basis for this two-trial rule is less absolute than it appears, and in practice FDA has regularly approved drugs on the basis of a single trial.
The Statistical Argument
If two independent trials each require a one-sided p-value below 0.025 to succeed, and both just reach that threshold, the combined evidence has a p-value of approximately \(0.025^2 = 0.000625\). This is the actual combined evidence threshold the two-trial rule demands from a sponsor who runs two trials, each at exactly \(p < 0.025\). The implication: a sponsor who designs a single trial to achieve \(p < 0.000625\) presents roughly equivalent evidence to a two-trial program, at potentially lower overall sample size.
The efficiency arithmetic is concrete. Two trials each powered at 90% give the development program \(0.90 \times 0.90 = 81\%\) power. A single trial designed to the combined threshold can achieve equivalent program power with approximately 80% of the total sample size of the two-trial program, because the experiment is unified rather than split. The two-trial rule also creates a classification problem: a program where trial 1 achieves \(p = 0.001\) and trial 2 achieves \(p = 0.026\) technically “wins one and loses one,” yet the combined evidence (roughly \(p = 0.003\)) substantially outperforms two trials each achieving \(p = 0.024\) (combined roughly \(p = 0.006\)). Analyzed by pooling the evidence rather than by pass/fail of each trial, the evidence ordering reverses.
When FDA Accepts a Single Trial
FDA has codified the single-trial exception in statute (FDAMA 1997, 21 U.S.C. § 355(d)) and in guidance (U.S. Food and Drug Administration 2019). A substantial share of recent approvals have been based on a single trial together with confirmatory evidence rather than two fully independent replications.
The conditions that support a single-trial submission are:
- Hard clinical endpoint. Survival, cardiovascular death, or other endpoints where the biological connection to patient outcome is unambiguous. Surrogate endpoints carry more risk of false-positive results that would not replicate.
- Strong p-value. The trial must achieve a substantially lower threshold than the two-trial rule’s conventional 0.025, reflecting the equivalent combined-evidence standard. FDA interactions for specific programs determine the pre-agreed threshold.
- Large multicenter enrollment. Demonstrates generalizability and reduces the risk that results are site-specific.
- Internal consistency. Pre-specified subgroup analyses should show that the overall result is not driven by one atypical subgroup, effectively providing the within-trial replication that two trials provided across trials.
- Confirmatory mechanistic or surrogate evidence. Biological plausibility and supportive Phase II data strengthen the case.
The Breadth Tradeoff
The genuine statistical cost of the one-trial approach is breadth. When sponsors run two trials in different but related patient populations, approval covers both, and labeling reflects evidence in each. A single trial in one population yields a label limited to that population, with off-label use carrying no safety or efficacy guarantee.
For sponsors designing around the one-trial standard, this argues for enrolling a broader population within the single trial and pre-specifying subgroup analyses that function like a basket, demonstrating that the effect is consistent across the range of patients who will use the drug in practice. A narrow initial label followed by post-marketing expansion trials is a common outcome when a single pivotal trial supports the first approval. The breadth question bears directly on labeling negotiations with FDA at the pre-BLA meeting, and sponsors should resolve the population scope before the pivotal trial is designed.
29.8 International Regulatory Pathways
While the U.S. NDA/BLA process is a central reference point, sponsors pursuing global registration must navigate parallel pathways in other major markets.
European Medicines Agency (EMA). The centralized procedure allows sponsors to submit a single Marketing Authorization Application (MAA) to obtain a marketing authorization valid in all EU/EEA member states. The centralized procedure is mandatory for certain product categories (biotechnology-derived products, orphan medicines, advanced therapy medicinal products) and optional for other innovative therapies. The Committee for Medicinal Products for Human Use (CHMP) conducts the scientific assessment, with a standard timeline of 210 active review days (European Medicines Agency 2022). Sponsors may also use decentralized or mutual-recognition procedures for products not requiring centralized authorization.
Pharmaceuticals and Medical Devices Agency (PMDA, Japan). Japan’s regulatory pathway involves the J-NDA (new drug application) submitted to PMDA, which conducts both the scientific review and GMP inspections. Japan historically required local clinical data, and bridging studies under ICH E5 remain common to demonstrate that foreign data apply to the Japanese population. However, Japan’s participation in ICH and acceptance of multi-regional clinical trial data under ICH E17 has increasingly enabled global submissions.
National Medical Products Administration (NMPA, China). China’s regulatory framework has undergone substantial reform since 2017, including acceptance of international multi-center clinical trial data for registration, elimination of the mandatory clinical trial import license delay, and alignment with ICH guidelines (China became an ICH member in 2017). These reforms have contributed to China’s emergence as a major clinical trial market (see Chapter 6). The NMPA still generally requires inclusion of Chinese patients in pivotal trials, and local regulatory expertise remains essential for navigating evolving requirements.
ICH harmonization and multi-regional submissions. The Common Technical Document (CTD/eCTD) format, maintained by ICH (M4), enables sponsors to prepare a single core dossier (Modules 2–5) for submission across ICH member regions, adapting only Module 1 for region-specific administrative requirements. ICH E17 provides the scientific framework for designing multi-regional clinical trials that can simultaneously support regulatory decisions in multiple jurisdictions, reducing the need for duplicative regional studies while preserving the ability to assess consistency of treatment effects across populations (International Council for Harmonisation 2017, 2016).
Regulatory obligations continue well beyond the initial approval. Sponsors are often required to conduct post-marketing requirements (PMRs) or agree to post-marketing commitments (PMCs), which can involve additional safety studies or trials in specialized populations. For therapies with significant identified risks, a Risk Evaluation and Mitigation Strategy (REMS) may be mandated to ensure safe use through communication plans or restricted distribution (U.S. Food and Drug Administration 2024b). Throughout the product’s marketed life, sponsors must monitor safety and submit periodic reports to regulatory authorities, confirming that the benefit-risk balance remains favorable as real-world experience accumulates.